II. EDA of BSSeq data generated from non-model organism in different conditions
BSXplorer allows for the categorization of regions based on their methylation level and density. This is done by assuming that cytosine methylation levels follow a binomial distribution, as explained in Takuno and Gaut’s research (please refer to [1, 2] https://doi.org/10.1073/pnas.1215380110 for details). The genes are then divided into three categories, BM (body-methylated), IM (intermediately-methylated) and UM (under-methylated), based on their methylation levels in the CG context using the following formula.
The same rationale may be applied to other methylation contexts, as BSXplorer can produce \(P_{CHG}\) and \(P_{CHH}\) for CHG sites and CHH sites, respectively.
[1] Takuno S, Gaut BS. Body-Methylated Genes in Arabidopsis thaliana Are Functionally Important and Evolve Slowly. Mol Biol Evol. 2012;29:219–27.
[2] Takuno S, Gaut BS. Gene body methylation is conserved between plant orthologs and is of evolutionary consequence. Proc Natl Acad Sci. 2013;110:1797–802.
Categorisation with console script
Firstly, we will demonstrate the categorization of regions and their visualization using a console script.
bsxplorer-category -o TE_Cat_Report --dir TE_Cat -u 50 -d 50 -b 100 -S 10 --ticks \\-500bp \\ Body \\ \\+500bp -C 0 -V 50 -H 50 -q .75 --save_cat te_conf.tsv
A user can obtain a complete list of parameters by using the command bsxplorer-category –help. The configuration file has the following structure
Header should NOT be included in real config file.
sample group |
Path to report |
Path to genome |
Flank length |
Minimal length |
Region_type |
|---|---|---|---|---|---|
Misugi_mock |
DRR336466.CX_report.txt.gz |
TE.gff |
500 |
0 |
match |
Misugi_mock |
DRR336467.CX_report.txt.gz |
TE.gff |
500 |
0 |
match |
Misugi_infected |
DRR336468.CX_report.txt.gz |
TE.gff |
500 |
0 |
match |
Misugi_infected |
DRR336469.CX_report.txt.gz |
TE.gff |
500 |
0 |
match |
The bsxplorer-category command imports Bismark’s cytosine report file and performs a binomial test
to determine the statistical significance of methylation at each site.
The command uses the estimated error rate to conduct the test and generates corresponding p-values,
which can be exported using the API. Additionally,
the tool imports genomic annotation for each region in every context and calculates the
PCG (or \(P_{CHG}\) or \(P_{CHH}\)) value.
Finally, it filters and classifies the regions into UM, BM and other classes based on the results obtained.
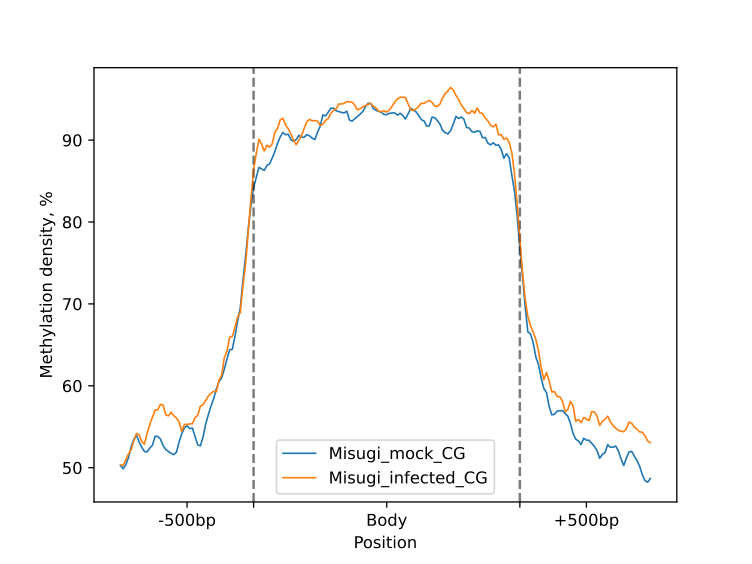
The following plots were generated for the BM and UM regions (CG-context) as part of the HTML-report.
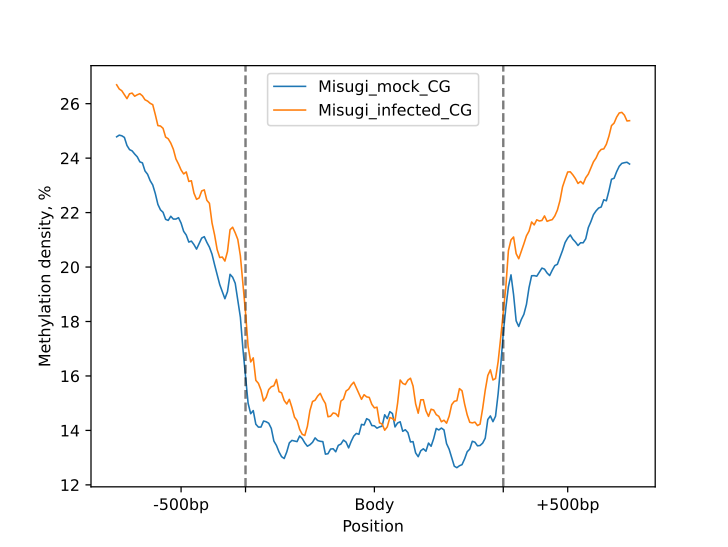
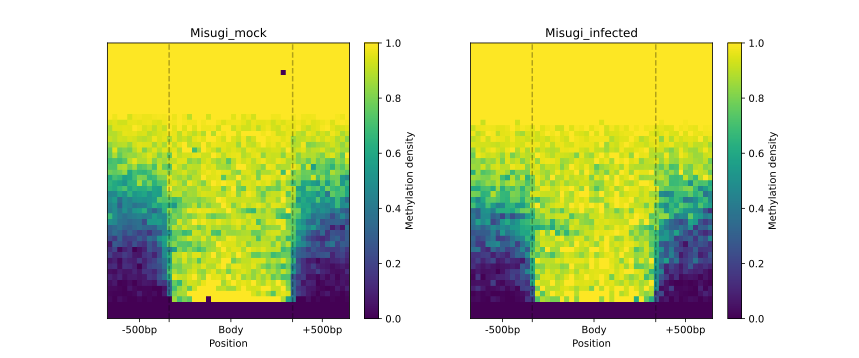
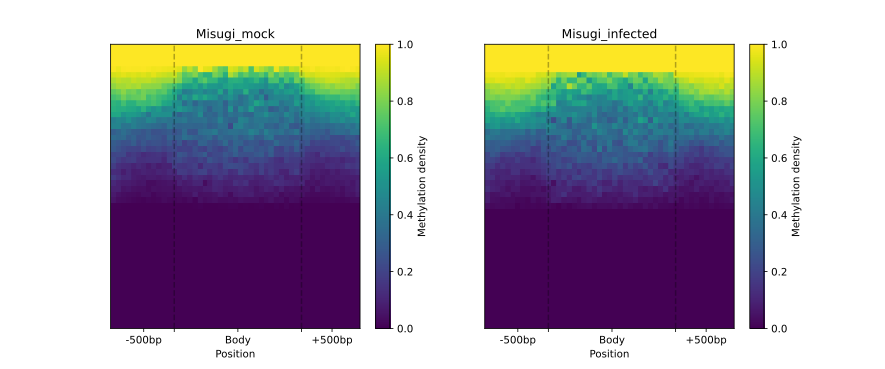
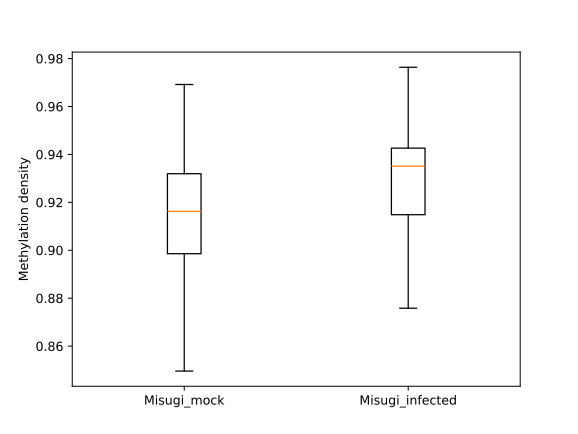
Categorisation with Python API
The API version of the above command string is presented in the code snippet below:
annot = bsxplorer.Genome.from_gff("TE.gff")
annot = annot.other(region_type="match", flank_length=500, min_length=200)
mock_merged = merge_replicates(
["DRR336466.CX_report.txt.gz", "DRR336467.CX_report.txt.gz"],
report_type="bismark"
)
inf_merged = merge_replicates(
["DRR336468.CX_report.txt.gz", "DRR336469.CX_report.txt.gz"],
report_type="bismark"
)
p_value_kwargs = dict(
genome=annot,
methylation_pvalue=.05
)
mock_binom = bsxplorer.BinomialData.from_report(
mock_merged.name, report_type="parquet", min_coverage=2
)
mock_pstat = mock_binom.region_pvalue(**p_value_kwargs)
inf_binom = bsxplorer.BinomialData.from_report(
inf_merged.name, report_type="parquet", min_coverage=2
)
inf_pstat = inf_binom.region_pvalue(**p_value_kwargs)
categorise_kwargs = dict(
context="CG", p_value=.05, min_n=5
)
# Returns tuple with (BM, IM, UM) ids
mock_cat = mock_pstat.categorise(**categorise_kwargs, save="mock")
inf_cat = inf_pstat.categorise(**categorise_kwargs, save="mock")
metagene_kwargs = dict(
genome=annot,
up_windows=50, body_windows=100, down_windows=50,
)
mock_metagene = bsxplorer.Metagene.from_binom(mock_binom.path, **metagene_kwargs)
inf_metagene = bsxplorer.Metagene.from_binom(inf_binom.path, **metagene_kwargs)
# e.g. for CG BM
metagenes = bsxplorer.MetageneFiles([
mock_metagene.filter(context="CG", genome=mock_cat[0]), inf_metagene.filter(context="CG", genome=mock_cat[0])
], ["Mock", "Infected"])
tick_kwargs = dict(
major_labels=["", ""],
minor_labels=["-500bp", "Body", "+500bp"]
)
metagenes.line_plot(smooth=10).draw_mpl(**tick_kwargs)
metagenes.heat_map(50, 50).draw_mpl(**tick_kwargs)
For detailed explanation of used methods and parameters see:
Conditions comparison
Using BSXplorer, the following section displays a practical approach to presenting these elements.
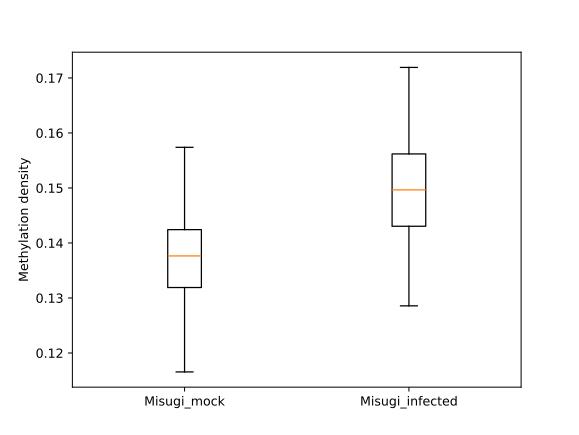
The control and infected samples are referred to as “mock” and “inf” respectively.
Here we assume that \(P_{CG}\) statistics which enables UM and BM classification has been computed in the previous
steps of analyses (by --save_cat option of bsxplorer-category or save parameter of RegionStat.categorise()).
After importing the bisulfite data for the transposons (Misugi_mockCG_UM.tsv and Misugi_infectedCG_BM.tsv),
the sets are intersected, matched with the annotation file,
and the resulting patterns are visualized and compared to other regions.
Here we will use Polars (https://pola.rs/) module for DataFrames manipulation.
import polars as pl
# Read mock_UM and inf_BM TE IDs
mock_um = pl.read_csv("Misugi_mockCG_UM.tsv", separator="\t", has_header=False)[:, 3].to_list()
inf_bm = pl.read_csv("Misugi_infectedCG_BM.tsv", separator="\t", has_header=False)[:, 3].to_list()
# Intersect TE IDs from two groups
mock_inf_up = list(set.intersection(set(mock_um), set(inf_bm)))
# Load annotation file
te = bsxplorer.Genome.from_gff("TE.gff").other("match", 0, flank_length=500)
The metagenes are constructed using the mean statistics with the sumfunc="mean" parameter.
args = dict(genome=te, up_windows=50, body_windows=100, down_windows=50, sumfunc="mean")
# Read replicates metagenes
metagene_mock = bsxplorer.MetageneFiles.from_list(["DRR336466.CX_report.txt.gz", "DRR336467.CX_report.txt.gz"], labels=["mock-1", "mock-2"], **args)
metagene_inf = bsxplorer.MetageneFiles.from_list(["DRR336468.CX_report.txt.gz", "DRR336469.CX_report.txt.gz"], labels=["inf-1", "inf-2"], **args)
# Merge biological replicates
metagene_mock = metagene_mock.merge()
metagene_inf = metagene_inf.merge()
The .merge() method of the MetageneFiles class is used to combine biological replicates.
The following code snippet creates a clustergram for a selected subset of genomic regions of interest using the
.dendrogram() function.
# Construct a MetageneFiles object to visualize dendrogram
dendro_metagene = bsxplorer.MetageneFiles([metagene_mock.filter(id=mock_inf_up), metagene_inf.filter(id=mock_inf_up)], ["mock_up", "inf_up"])
dendro_metagene.dendrogram(q=0)
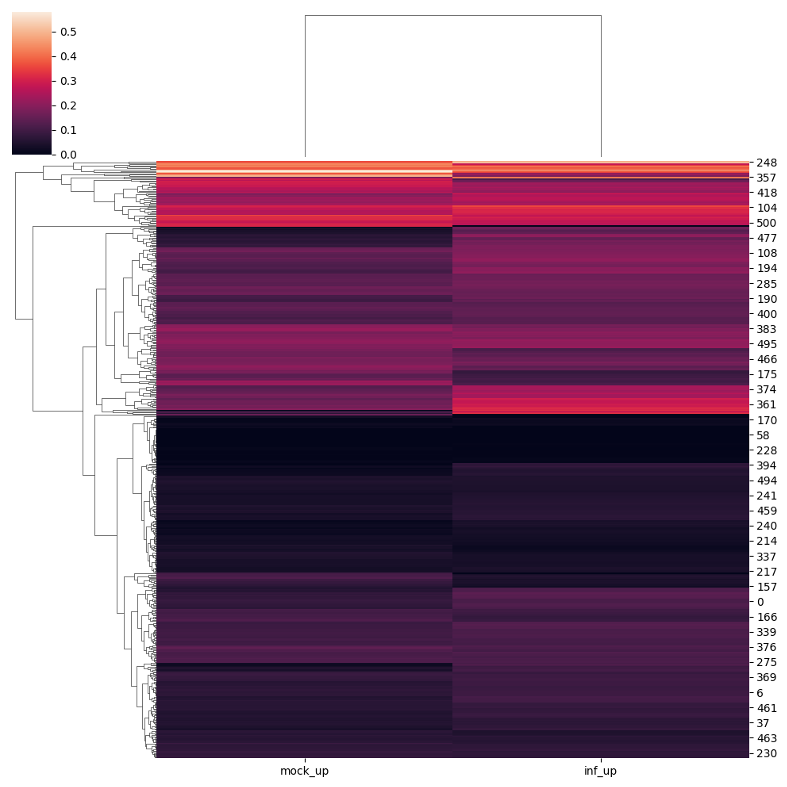
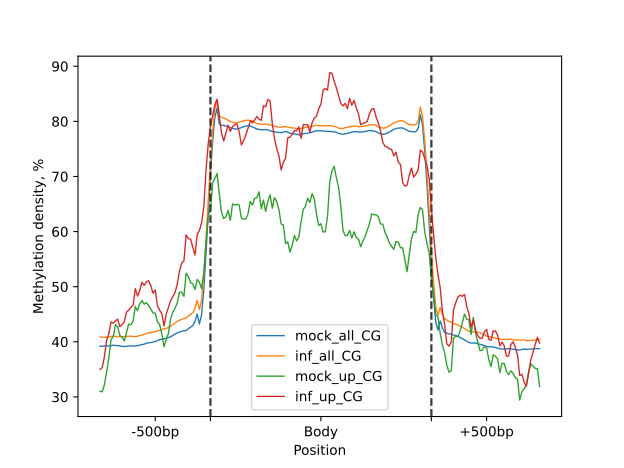
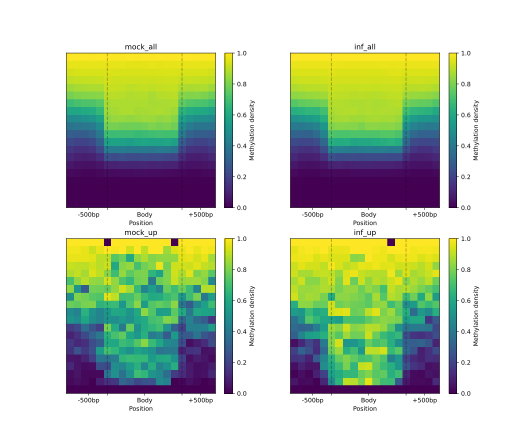
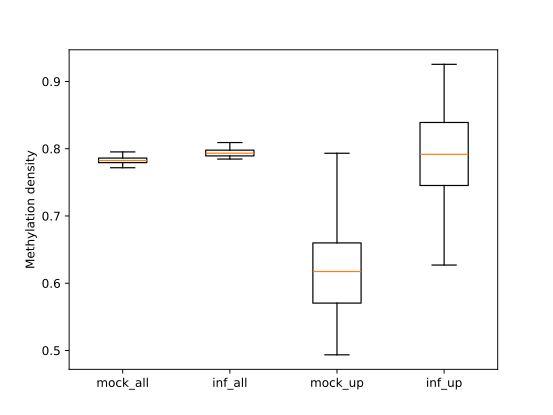
To visually compare the UM and BM regions that contain transposable elements with the remaining genomic regions that
exhibit UM or BM patterns, we create a MetageneFiles object. This object enables us to generate line and box plots, as
well as heatmaps that display methylation levels in the aforementioned classes.
metagenes = bsxplorer.MetageneFiles(
[metagene_mock, metagene_inf, metagene_mock.filter(id=mock_inf_up), metagene_inf.filter(id=mock_inf_up)],
["mock_all", "inf_all", "mock_up", "inf_up"]
)
filtered = metagenes.filter(context="CG")
ticks = {"major_labels": ["", ""], "minor_labels": ["-500bp", "Body", "+500bp"]}
filtered.line_plot(smooth=10).draw_mpl(**ticks)
filtered.heat_map(20, 20).draw_mpl(**ticks)
filtered.trim_flank().box_plot().draw_plotly()