Genome
Reference
Storing and manipulating annotation data. |
|
Class for performing logFC enrichment of specified regions over genome. |
|
Class for storing and visualizing enrichment results. |
Usage
BSXplorer offers functionality to align one set of regions over another. Regions can
be read either with Genome or initialized directly with
polars functionality
(DataFrame need to have chr, start and end columns).
To align regions (e.g. define DMR position relative to genes) or perform the enrichment of regions at these
genomic features against the genome background use Enrichment.
import bsxplorer as bsx
# If you want to perform an ENRICHMENT, and not only plot
# the density of metagene coverage, you NEED to use .raw() method
# for genome DataFrame.
genes = bsx.Genome.from_gff("path/to/annot.gff").raw()
dmr = bsx.Genome.from_custom(
"path/to/dmr.txt",
chr_col=0,
start_col=1,
end_col=2
).all()
enrichment = bsx.Enrichment(dmr, genes, flank_length=2000).enrich()
Enrichment.enrich() returns EnrichmentResult, which stores enrichment
statistics and coordinates of regions which have aligned with
genomic features. The metagene coverage with regions
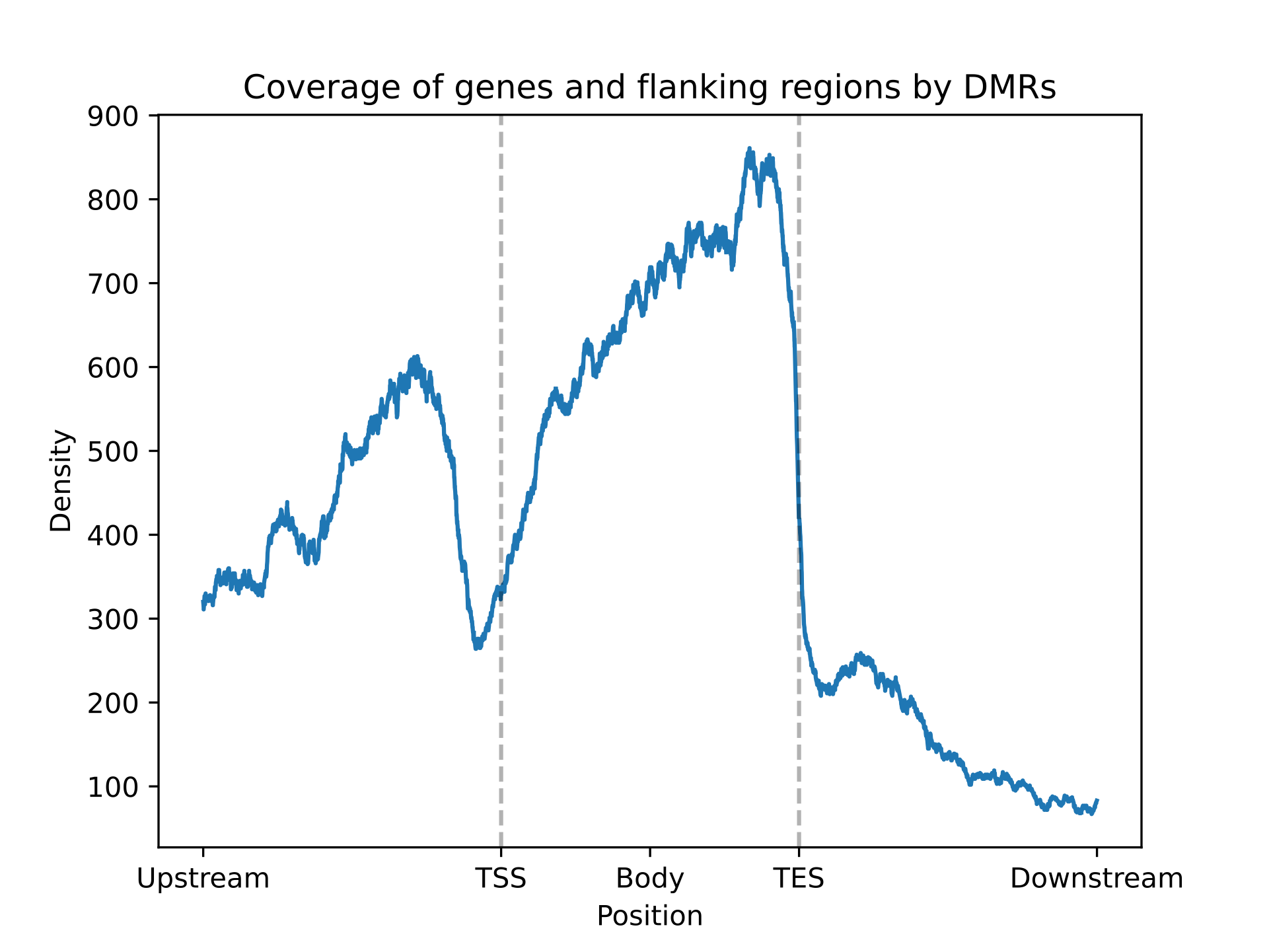
can be plotted via EnrichmentResult.plot_density_mpl() method.
fig = enrichment.plot_density_mpl()
Example of resulting image:
Enrichment statistics can be accessed with EnrichmentResult.enrich_stats
or plotted with EnrichmentResult.plot_enrich_mpl()